With all phenomena in the Life Sciences there are two major questions scientists ask: (1) what is the underlying mechanism (how?) and (2) what is its biological significance (why?). All eukaryotic cells replicate their DNA in a specific temporal sequence but both the mechanism of this “replication timing” program and its biological significance remain a mystery. Faithfully duplicating our DNA is arguably the most fundamental thing that our cells do, and mistakes in this process are major drivers of disease, yet we understand very little about its regulation. Replication timing is clearly related to the 3-dimensional organization of chromosomes such that early and late replicating DNA are spatially segregated from each other in the nucleus (Figure 1) and the timing program is developmentally regulated at the level of large segments of chromosomes. The order in which these segments replicate is disrupted in many diseases, including all cancers. However, we still have no conclusive understanding of why replication takes place in the particular order it does, or why it is different in each of the tissues of our body.
The primary motivation for our research is that these units that we call “replication domains” provide us with a molecular handle into the higher order structural and functional organization of chromosomes. We hypothesize that, since it is not just DNA that replicates but the entire structure of chromatin and its 3D organization, controlling where and when replication takes place can serve as a convenient means for cells to maintain stable epigenetic states and to change those states during cell fate transitions. The work in our laboratory can be divided into the following major areas:
1. Developmental Regulation of DNA Replication:

All cells contain the same genetic information (DNA) but package it with proteins into “chromatin” in characteristic ways that define each cell type. Chromatin is dismantled and re-assembled during each round of DNA replication, and we have discovered that the temporal order in which segments of DNA are replicated changes as stem cells turn into different cell types and is mis-regulated in disease. Understanding how to manipulate this packaging process may help us engineer different cell types, a central goal in stem cell therapy. Replication proceeds via the synchronous firing of clusters of replication origins encompassing domains of several hundred kilobases (Figure 2). These “replication domains” coincide with structural and functional domains of chromosomes and replicate in a defined temporal order during S-phase. Since chromatin is assembled at the replication fork, and since different types of chromatin are assembled at different times during S-phase, it makes sense that replication would be an important regulatory event at which to assemble different types of chromatin in different cell types, but testing this hypothesis has been difficult. Many studies have correlated changes in replication timing to changes in gene expression in different cell lineages and in cancer but none have been able to address the intermediate states that accompany these changes. Mechanistic studies require a system in which these changes can be elicited with sufficient synchrony and homogeneity as to permit biochemical and molecular analyses. Using differentiation systems modeling both mouse and human early development, we detect dynamic changes in replication timing that affect at least half of the genome, some occurring within a single cell cycle (Figure 3). Since these dynamic replication changes, unlike other functional properties of chromosomes, are regulated at the level of large chromosomal domains, our studies probe uncharted mechanisms in gene regulation.

Our working hypothesis is that changes in replication timing during differentiation change the types of proteins and RNA that assemble with the DNA into chromatin and since large (half megabase) segments of chromosomes replicate as units, these changes occur across large chromosome domains. These chromatin changes would then modulate the responsiveness of genes during stem cell commitment.
Together, this body of work has provided a watershed of information that has upheld or refuted longstanding hypotheses about replication timing and generated many new hypotheses that are now testable with the systems that we have developed. We are taking both genomics approaches to evaluate the significance of replication timing to cell fate changes during differentiation, and more targeted approaches to elucidate the mechanisms relating replication timing and gene expression changes at specific gene loci.
For example, taking advantage of the information in the datasets linked to the lineage diagram in Figure 3, we were able to perform network analyses that link the expression of specific transcription factors to sets of domains throughout the genome that are regulated during liver and pancreas development and identified novel interactions between these transcription factors and their targets that may regulate the developmental changes in replication timing (https://doi.org/10.1101/186866).
2. Regulation of Replication Timing During the Cell Cycle:
Studies of DNA replication in mammalian cells suffer from a lack of systems with which to approach the problem at the molecular level. Cis-acting sequences that function as replication origins in mammalian cells have not been identified and the mechanisms that regulate where and when origins will fire during S-phase remain a mystery. Over the last 25 years we have taken a variety of approaches to this problem, revealing several discrete steps during early G1-phase that establish a spatial and temporal program for replication:
Pre-Replication Complex (pre-RC) Assembly: First is the assembly of pre-replication complexes (pre-RCs). Knowledge of this process has been greatly aided by the identification of several proteins that bind to replication origins in yeast and the identification of homologues to these proteins in higher eukaryotes. We have shown that the mammalian homologues to these proteins bind to chromatin very tightly during telophase and render newly assembled nuclei fully competent to replicate in cell-free extracts lacking these proteins.
The Replication Timing Decision Point (TDP): Shortly after the assembly of pre-RCs, a replication timing program is established that determines the order in which chromosomal domains will be replicated (Timing Decision Point; TDP).
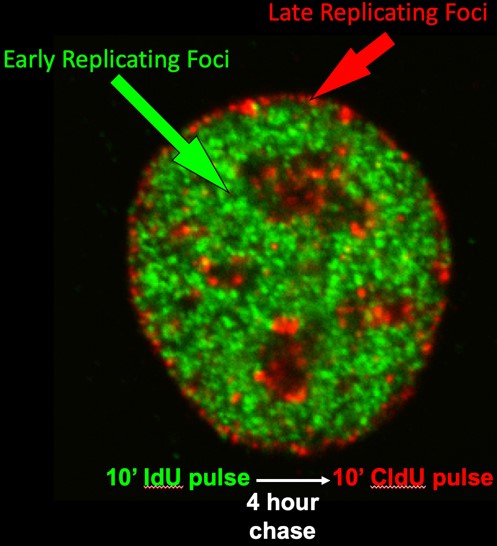
Early cytogenetic studies by our group and others demonstrated that DNA synthesis takes place in discrete punctate foci within the cell nucleus and that foci replicating at different times during S phase are located in distinct compartments of the nucleus. We and others have shown that these foci are visual representations of stable structural units of chromosomes that replicate at specific times. We have demonstrated that the replication-timing program is established within 2 hours after the cell nucleus is formed after each mitosis, a time we called the Timing Decision Point (TDP), and that the re-positioning and anchorage of these foci within the nucleus occurs at the same time. The TDP precedes the time during G1-phase at which the sites for initiation of replication are established (Origin Decision Point; ODP), a regulatory point we discovered back in the 1990s (https://science.sciencemag.org/content/271/5253/1270.long). In fact, we can create conditions in which replication proceeds in the proper temporal order but initiates at random sites, showing that the two are regulated independently. We later demonstrated that determinants for replication timing are lost during S phase, despite maintenance of the 3D organization of chromatin, so we can say that the 3D organization is not sufficient to dictate replication timing. Thus, a central question for us is, does structure dictate function or does function dictate structure?
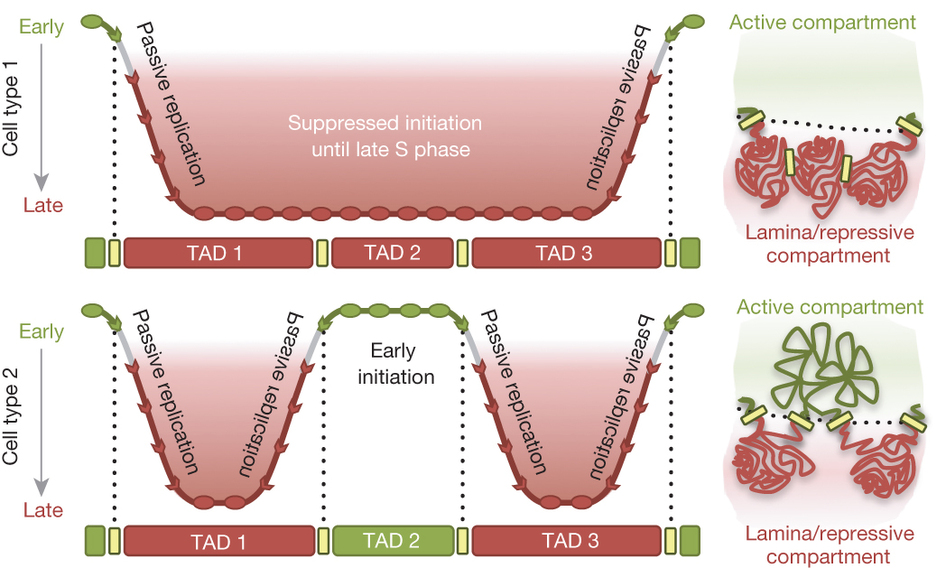
Taken together, our findings have led us to propose the “Replication Domain Model” (Figure 4).
Our early work, mostly using microsopy, showed that replication foci (Figure 1; discussed above) are stable units of chromosome structure consisting of multiple coordinately activated replicons that retain the punctate replication pulse label for many cell generations. We showed that replication foci serve as sites of replication protein assembly and disassembly in a temporal sequence that can be uncoupled from DNA synthesis itself. We found that foci in different compartments have different chromatin composition and proposed that, since chromatin is assembled at the replication fork, organization of the genome into coordinately replicated domains could facilitate rapid domain-wide chromatin changes.
Using genomics, we discovered that replication timing consistently changes in units of 400-800 kb, defining molecular coordinates for replication domains. We showed that domain boundaries are functionally relevant in that they can confine the effects of rearrangements. We demonstrated that replication domains correspond to topologically associating domains (TADs) measured by chromatin conformation capture (Hi-C) and their higher order folding corresponds to replication timing such that domains that contact each other replicate at similar times (https://www.nature.com/articles/nature13986). Both the TADs and the higher order 3D organization is re-established shortly after the nucleus is reformed after mitosis and coincident with the TDP (above), when replication timing is established. In summary, our work shows that the way the genome is organized in 3D space has important functional consequences and provides a unifying model for genome organization that some have generously likened to the discovery of the nucleosome. We propose that anchorage of chromatin could create scaffolds that seed the assembly of sub-nuclear compartments of different molecular composition, a model that has become popular to explain many structure-function relationships in the nucleus. We have also identified a candidate protein (Rif1) that has properties consistent with a cell cycle regulated factor important for both replication timing regulation and 3D organization of chromatin.
3. Discovery of ERCEs:
The holy grail of this field since the discovery of a replication timing program in 1960 has been the identification of the control elements in chromosomes for this temporal regulation. Many investigators have tried and failed to identify them, leading scientists to propose that replication timing is not regulated by DNA sequences. With the advent of CRISPR, allowing us to make large numbers of DNA mutations in a single “replication domain”, we identified specific DNA elements that are both necessary (required in their chromosomal location) and sufficient (active in other chromosome locations) for a domain to replicate early during S phase. Consistent with our “replication domain” model, these elements are also required for the 3D structure of the chromosome domain and for localization of the domain to its sub-nuclear compartment and, we believe independently, for transcription of all genes within the domain. Consistent with our network analyses pointed out in section 1 above, ERCEs are also sites of co-occupation of transcription factors that are essential for the identity of embryonic stem cells, the cell type in which we identified these ERCEs.
This is a major breakthrough in our field. Working out the mechanisms by which these elements control structure and function of entire chromosome domains will drive research in the lab for many years to come. (https://www.cell.com/cell/pdf/S0092-8674(18)31561-7.pdf)
4. Solving the mystery of human origins:
Another one of the great holy grails in the DNA replication field is to find the origins of replication. We believe that ERCEs regulate the time at which an entire chromosome domain is ready to replicate but they are not the sites where replication begins, the “origins”. Scientists have argued whether origins are specific sites or whether replication can initiate at any site in the genome at random. Most of these scientists ignore one body of evidence or another.
We believe, based on taking ALL the evidence into account, that humans (and other organisms with large genomes) have a great deal of flexibility as to where replication can initiate and that these sites are selected stochastically (stochastic is not random, but rather with each of the potential sites of initiation being used with a different probability). As a result, each cell in a population is using a different cohort of initiation sites. Thus, we believe, all the methods that have been used to map origins that pool millions of cells to map origins are getting only an average picture, with any one site being used as an initiation site very rarely so being very difficult to detect. This has resulted in datasets that do not agree, fuelling the polarization of the field.
The solution to this problem, and the test of our hypothesis, is to measure replication initiation on single DNA molecules, one at a time, in order to actually measure how frequently replication initiates at all sites across the genome. We predict that many sites, but not all, will be used to initiate replication and that the frequency of usage will vary widely across the genome.

We are taking two approaches to this challenge. One is to take advantage of very new technology that can stretch individual purified DNA fibers and read their map positions at an unprecedented rate – we add to that a label to tell us where replication has initiated (Figure 5). We believe that this is revolutionary technology that is poised to change how we map and study DNA replication and we intend to invest significant resources into this new approach. (https://doi.org/10.1101/214841).
In the second approach, we are trying to develop methods to map the positions of protein binding on single chromosomes. This is in the development state, but if successful, we will be able to map the sites where pre-replication proteins bind during G1 phase. This will allow us to map the specificity and degree of stochastic positioning of these “markers” that are necessary for replication to initiate and relate that to the actual sites of initiation mapped on the purified DNA fibers. This is the work of Dan Bartlett in the lab, currently unpublished.
5. Replication timing and disease:
By comparing normal and diseased cells, we can identify alterations in replication timing that are specific to disease. Two examples are as follows:
By comparing normal skin cells from individuals of widely different ages, from fetal to 96 years old we did not find significant differences in replication timing. However, premature aging diseases, which can arise from mutations in very different genes, have been puzzling because there has been no molecular commonality between these different genetic origins of the disease even though it gives rise to the same clinical phenotype. Comparing skin cells from normal individuals of different ages and patients with premature aging disease from different genetic origins revealed a set of common replication timing abnormalities: the first molecular marker of these diseases. Moreover, by converting these cells into embryonic stem cell-like cells and then re-differentiating them back to skin cells, we could re-capitulate human embryogenesis in a dish and demonstrate which of these replication timing abnormalities occur first during human development. This identified a specific gene, TP63, as a central player in premature aging disease. How the misregulation of TP63s replication timing can dramatically accelerate the process of aging is now an area of active pursuit for Dr. Rivera-Mulia in his new position at U. of Minnesota. (https://www.pnas.org/content/114/51/E10972.short)
B-lineage acute lymphocytic leukemia (B-ALL) is the most common childhood malignancy, yet we still do not understand the molecular mechanisms that lead to the genesis of this disease and there are no known strategies for its prevention. We demonstrated that pediatric B-ALL cells show alterations in replication timing from non-leukemic human B cells. Some of these alterations are linked to particular sub-types of B-ALL defined by their hallmark mutations and resemble features of specific stages of normal human B-cell differentiation suggesting that different subtypes my derive from cells arrested at these different stages. Intriguingly, some of these features were linked to whether the patients remained in remission or relapsed. Thus, RT has the potential to provide a new genre of biomarkers for diagnosis. In addition, RT reports on an undeveloped aspect of chromosome biology that is altered in B-ALL. Hence, studies of the cellular origin and mechanistic determinants of these RT alterations will provide novel insights into the underlying mechanisms of B-ALL. (https://doi.org/10.1101/549196)
6. The biological significance of replication timing: driving changes in epigenomic state to facilitate cell fate transitions.
In collaboration with Sara Buonomo (U. Edinburgh), we identified the first trans-acting factor whose depletion causes a nearly complete elimination of the replication timing program, allowing us to finally ask questions about the function of replication timing. We then demonstrated that disruption of replication timing leads directly to widespread, replication-dependent, re-distribution of histone post-translational modifications and alterations in genome architecture that require replication, demonstrating that replication timing is necessary for the maintenance of chromatin (epigenetic) states. It is now imperative to determine whether the mechanisms by which replication timing contributes to the epigenome is through the assembly of specific chromatin states at particular times and locations in the nucleus. (DOI: 10.1126/science.aba5545)
7. Oncogenic pathway-induced fragile sites: a new paradigm for understanding genome instability in cancer
Funded by NIH/NCI grant R01CA270335 to MPI David Gilbert, MPI Batsheva Kerem, co-I Kadir Akdemir, co-I Nicola Neretti
One of the grand challenges in cancer research is the vast heterogeneity in responsiveness to treatment for different cancer types. We have made great strides in treating some cancers, while the prognosis for others remains dismal. Large scale whole genome sequence (WGS) analyses have identified “breakpoint signatures” of different cancer types that presumably reflect heterogeneity in their underlying disrupted pathways but, drug-gable targets have emerged only in cases where the breaks create oncogenic fusion proteins. The ability to predict disrupted pathways using WGS would be highly impactful for cancer diagnosis and treatment. Chromo-some fragile sites (FSs) manifest as gaps and breaks in metaphase chromosomes when cultured cells experi-ence replication stress. However, many FSs are not cancer-type specific and many breakpoint hotspots in cancer are not known FSs, which has severely limited the impact of the FS field. Since cultured cells offer the potential to prospectively dissect mechanisms initiating chromosome breaks and to track their expansion into complex structural variation, there is a critical need to make these in vitro systems more cancer relevant. Our longterm goal is to establish a sustained research strategy that can predict which known pathways are disrupt-ed in a given cancer type from their breakpoint patterns. The overall objective of this proposal is to reveal mechanisms by which perturbation of cancer-relevant cellular pathways produce pathway-specific patterns of FSs in cell culture and determine whether those patterns can be identified in cancers. Our central hypothesis is that the power of in vitro systems to dissect mechanisms leading to cancer breakpoint signatures will become evident only when shifted away from chemically-induced FSs towards FSs induced by perturbing known can-cer-relevant pathways. We have shown that overexpression of different oncogenes leads to oncogene-specific spectra of FSs and preliminary data suggest some of the mechanisms by which this occurs. Our rationale is that understanding cancer-relevant mechanisms that specify FSs will fill the gap in linking in vitro FSs to cancer breakpoints. This would be a major step toward a strategy to predict disrupted pathways from cancer WGS da-ta, thereby suggesting treatments for previously intractable cancers. Aim1 will use high throughput/resolution replication and Oligopaints assays to identify downstream mechanisms distinguishing which of many sites of oncogene-induced replication delay manifest as FSs. Aim2 will elucidate upstream mechanisms causing replica-tion delays and FS at specific sites. Aim3 will map FSs at unprecedented resolution and mine tumor sequencing databases for signatures that match those of oncogene-specific FSs. This contribution will be significant be-cause the ability to identify affected pathways solely from breakpoint signatures would expose tumor-specific vulnerabilities for precision cancer medicine. The proposed research is innovative because it will rejuvenate the impact of FS research, closing the gap between in vitro systems and in vivo cancers while leveraging novel technologies to manipulate cells, map their signatures of genome instability and match them to cancer break-point signatures.
8. The 3D architecture of native human replisomes and the effect of replication stress on this architecture:
Funded by NIH/NHGRI grant R01HG010658
Considerable effort is underway to understand the role of 3D genome architecture in transcription but, little at-tention has been paid to its role in DNA replication. DNA replication involves the complete disassembly and re-assembly of chromatin, providing a unique window into principles of genome architecture. Also, DNA and RNA polymerase collisions create R-loops that are a major source of genome instability. Knowledge of how DNA rep-lication navigates through complex genome architectures and is coordinated with transcription is fundamental to our understanding of chromosome biology and genome stability, yet we have little more than a rudimentary de-scription of replication in its native 3D context. At the root of this gap is the inherent heterogeneity of replication kinetics. Currently available maps of replication origins and fork polarities suffer from low signal-to-noise ratios due to the stochastic selection of different sites in each cell cycle from a high abundance of potential initiation sites, each of which has a different probability of firing. Single molecule methods have revealed this heterogenei-ty, but are extremely low throughput. Consequently, dispute over the specificity of origins in mammalian cells has persisted for decades. To move beyond the status quo there is a critical need for direct measurements of the locations of human origins, the polarities of forks, as well as their 3D proximity to each other and to ongoing transcription. AIM1 will deliver the first comprehensive, high confidence, single molecule maps of human replica-tion origins and fork kinetics that will address longstanding fundamental questions in DNA replication such as how origins are distributed, how initiation is coordinated within domains and how domains fire in a defined tem-poral order. AIM2 will produce the first 3D maps of in situ replisome complexes, including how 3D contacts and transcription are managed during replication. Finally, AIM3 will look at how close-range DNA interactions at ac-tive replication forks are coordinated across the genome and in single cells. This contribution will be significant because it will lead to a considerably deeper understanding of how DNA replication is orchestrated to preserve genome integrity and cell-type specific chromatin architectures. In doing so, this project also develops several highly innovative technologies that will open new horizons to study the 3D architecture of any process involving DNA synthesis (e.g. recombination, repair, genome stability and chromatin assembly).
9. Integrating Imaging and Omics by leveraging replication timing and the new telomere-to-telomere genome assemblies.
Collaborative project funded by NIH Common Fund grant UM1HG011593 to the labora-tories of Jian Ma, Andrew Belmont, Ting Wu, Frank Alber, Susanne Rafelski, David Gilbert, Nicola Neretti, Tom Misteli and Jason SwedlSwedlow
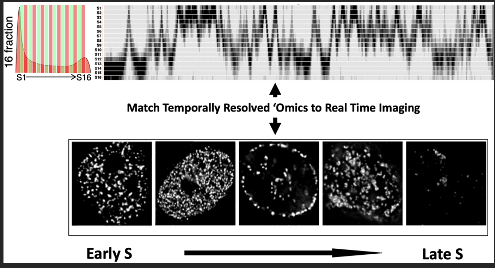
A major impediment to understanding the structure and function of cell nuclei is to integrate microscopy with genomic measurements of nuclear organization to understand how biological processes work in 4 dimensions (4D= space and time). DNA replication is an inherently 4D process that illuminates the spatial and functional organization of the genome as it unfolds over the course of S phase. This choreography, with internal, transcriptionally active euchromatin replicating first, followed by lamina- and nucleolar-associated facultative heterochromatin and finally constitutive heterochromatin is conserved from single celled ciliates to hydra, flies and vertebrates. This spatio-temporal program is developmentally regulated; in humans at least half of the genome changes its time of replication and sub-nuclear position during development, regulated in units that have the properties of Topologically Associated Domains (TADs). We have developed a high resolution Repli-seq protocol that identifies with high precision the temporal order in which genomic sequences replicate during the course of S phase (the 16 rows in the top panel of Figure 7). We and many others have also shown that tracking the locations of the replication fork protein PCNA illuminates the subnuclear locations where DNA synthesis is taking place in real time in living cells (bottom panel of Figure 7). We propose to link the spatio-temporal positions revealed by PCNA with the DNA sequences revealed by Repli-seq. This is a collaborative project between the laboratories of Jian Ma, Andrew Belmont, Ting Wu, Frank Alber, Susanne Rafelski, Nicola Neretti, Tom Misteli and Jason Swedlow.
10. Coordination of chromosome-wide replication timing, chromatin interactions, and monoallelic gene expression.
https://www.nature.com/articles/s41467-022-34099-7

Cancer genomes are characterized by numerous karyotypic abnormalities, including structural and whole chromosome numerical alterations. The single most significant factor associated with poor prognosis in solid tumors is an unstable genome, yet the tools to target cells with genome instability remain a significant challenge. While there have been many models proposed to explain genome instability in cancer cells, the underlying abnormalities remain largely unknown. Nearly 20 years ago, Matt Thayer (OHSU) discovered that 80% of cancers harbor a single chromosome that enters mitosis with an incompletely replicated and uncondensed chromosome and is highly unstable, resulting in breakages resembling chromothripsis (Figure 8). He has since discovered cis elements (ASynchronous replication and Autosomal RNAs; ASARs) required for timely replication and condensation of entire chromosomes. ASARs are lncRNAs (>200kb) that control chromosome-wide RT and are necessary to maintain structural stability across the chromosome in which they reside. Thus, disruption of ASARs eliminates the coordinated origin firing along chromosome pairs, giving rise to the phenotype seen in Figure 8. ASARs are that are mono-allelically expressed, non-spliced, Poly A—, display asynchronous replication and have a high L1 content. ASARs coat the chromosome from which they are expressed in cis, and their functional activity is mediated by L1 elements that are transcribed in the anti-sense orientation. Matt has undoubtedly uncovered a major regulator of chromosome structure and function; please appreciate this: a single 6 kb deletion can cause the chromosome-wide phenotype shown in Figure 8! I have followed Matt’s work since 2001 and I am excited that his work has reached a level where my lab can contribute. We have recently collaborated to identify 421 lncRNAs that share these “ASAR” characteristics. These 421 genes comprise ~4% of the genome, and we predict they are major regulators of chromosome homeostasis, yet they remain largely uncharacterized.

ASARs are thus likely to be essential cis-acting chromosomal elements controlling RT, monoallelic gene expression, mitotic chromosome condensation, 3D nuclear localization, chromatin interactions and structural stability, and likely interact in some way with ERCEs (Figure 9). Matt’s hypothesis is that ASARs recruit RNA binding proteins to coordinate replication chromosome-wide, likely affecting ERCEs. He has now shown that knockdown of proteins that bind to ASARs causes delayed condensation and replication of all chromosomes (https://doi.org/10.1101/2022.06.04.494840). Matt and I welcome trainees in either of our labs to work collaboratively on the biology of ASARs.
